Classification, diagnosis, and differential diagnosis of multiple sclerosis

Selected By
June Halper, MSN, APN-C, MSCN, FAAN
Why is this important to me?
Early and accurate diagnosis of MS is important because effective therapies are available, and if you receive early treatment, you are more likely to obtain a better outcome. MS needs to be diagnosed carefully because it can resemble other diseases. The ability of physicians to diagnose MS accurately is improving.
What is the objective of this study?
The authors reviewed the different subtypes of MS and how MS is distinguished from other diseases that may appear to be similar.
MS can be categorized into several different subtypes:
- Relapsing-remitting MS
- Most people with MS have this subtype.
- The relapsing-remitting disease course means that you experience periods of disease activity and symptoms, followed by periods of improvement.
- Diagnosis of this subtype requires the presence of typical symptoms and imaging results that show lesions in different places in your brain and/or spinal cord that developed at different times.
- At least two clinical attacks are needed for a diagnosis of relapsing-remitting MS
- Clinically isolated syndrome
- This is the initial presentation of symptoms that suggest demyelination (MS involves loss of myelin, the fatty substance that surrounds and protects nerve fibers)
- Clinically isolated syndrome is a pre-MS condition, and early treatment can prevent or delay conversion to MS.
- Higher levels of vitamin D are associated with a lower risk of developing MS if you have CIS.
- Radiologically isolated syndrome
- This subtype is diagnosed in people who have no symptoms that suggest MS but in whom imaging performed for another reason just happened to identify lesions that suggest MS.
- Around one-third of people with this subtype will develop MS-like disease within about 4 and a half years.
- Treatment varies due to a lack of definitive evidence.
- Primary progressive MS
- If you have this subtype, you will experience a progressive decline in your neurological function, rather than periods of worsening and recovery as in relapsing-remitting MS.
- Secondary progressive MS
- After 20 or so years with relapsing-remitting MS, about 40% of people will convert to secondary progressive MS.
- Similar to primary progressive MS, secondary progressive MS involves gradual worsening of your neurological function.
Distinguishing MS from other conditions:
MS resembles several other conditions in terms of symptoms and results from imaging studies. Several syndromes or symptoms suggest the possibility of MS. The details of clinical symptoms, imaging results, and spinal tap results and their combined results can help distinguish MS from another condition. Careful consideration of the following syndromes by a physician can help distinguish between suspected MS and suspicion of another condition:
- Spinal cord syndrome
- MS commonly presents with symptoms that suggest a problem in the spinal cord. Symptoms consistent with inflammation in the spinal cord (called “transverse myelitis”) related to MS include bladder and bowel problems and motor control problems. In contrast, paralysis of the lower limbs suggests another problem.
- Results from imaging and from a spinal tap can further distinguish MS-related spinal cord problems from non-MS spinal cord problems.
- Optic neuritis
- Optic neuritis is inflammation of the optic nerve.
- Acute problems in one eye and eye pain, which peak in a few days and recover in a few weeks, are consistent with MS. In contrast, problems in both eyes, much more rapid onset, or much slower onset suggest other problems. Lack of pain also suggests something else.
- Brainstem and cerebellum syndrome
- The presence of double vision (called “diplopia”) suggests an MS-related brainstem/cerebellum problem. Facial weakness and dizziness are also consistent with MS. Rapid onset of these problems suggests something else.
- Hiccups, vomiting, and nausea suggest another condition.
- Cognitive impairment
- Cognitive problems occur early in MS and are present in all subtypes. When cognitive problems are present along with other problems suggestive of MS, they can help confirm suspected MS. Cognitive problems alone are not enough to solidify a suspicion of MS.
- On imaging, certain shapes, sizes, and locations of brain and spinal cord lesions are characteristic of MS and can help confirm a suspected MS diagnosis. Other presentations suggest other conditions.
How did the authors study this issue?
The authors reviewed MS subtypes and characteristics that can help distinguish MS from other problems.
| SHARE: | |||||
Original Article
Classification, diagnosis, and differential diagnosis of multiple sclerosis
Ilana Katz Sand
Current Opinion in Neurology
Purpose of review
The increasing availability of effective therapies for multiple sclerosis as well as research demonstrating the benefits of early treatment highlights the importance of expedient and accurate multiple sclerosis diagnosis. This review will discuss the classification, diagnosis, and differential diagnosis of multiple sclerosis.
Recent findings
An international panel of multiple sclerosis experts, the MS Phenotype Group, recently revised the multiple sclerosis phenotypic classifications and published their recommendations in 2014. Recent research developments have helped improve the accuracy of multiple sclerosis diagnosis, especially with regard to differentiating multiple sclerosis from neuromyelitis optica spectrum disorders.
Summary
Current multiple sclerosis phenotypic classifications include relapsing-remitting multiple sclerosis, clinically isolated syndrome, radiologically isolated syndrome, primary-progressive multiple sclerosis, and secondaryprogressive multiple sclerosis. The McDonald 2010 diagnostic criteria provide formal guidelines for the diagnosis of relapsing-remitting multiple sclerosis and primary-progressive multiple sclerosis. These require demonstration of dissemination in space and time, with consideration given to both clinical findings and imaging data. The criteria also require that there exist no better explanation for the patient’s presentation. The clinical history, examination, and MRI should be most consistent with multiple sclerosis, including the presence of features typical for the disease as well as the absence of features that suggest an alternative cause, for a diagnosis of multiple sclerosis to be proposed.
INTRODUCTION
Since the introduction of interferon in 1993, ongoing research has yielded increasing availability of effective options for the treatment of multiple sclerosis. This, in combination with data suggesting the importance of early therapeutic intervention, highlights the critical nature of prompt and accurate multiple sclerosis diagnosis. When patients present with typical signs and symptoms and have imaging that is consistent with multiple sclerosis, the diagnosis can be relatively straightforward. However, when patients do not easily fulfill diagnostic criteria or when atypical clinical or imaging features are present, making the correct diagnosis may prove challenging even for an experienced neurologist. This review will illustrate the application of recent revisions to multiple sclerosis phenotypic classifications as well as the process for consideration of the diagnosis and differential diagnosis of relapsing and progressive forms of multiple sclerosis.

DEFINITIONS, CLASSIFICATION, AND DIAGNOSTIC CRITERIA
Multiple sclerosis is an inflammatory demyelinating disease affecting the central nervous system (CNS), thought to result from the interaction of genetic and environmental factors that remain only partially understood [1,2]. The pathogenesis of multiple sclerosis is also complex and incompletely understood, but the major principles underlying the disease seem to be inflammation and neurodegeneration. The previous classification scheme relied on the idea of the existence of distinct phenotypes dominated by either underlying inflammatory (relapsing-remitting) or neurodegenerative (progressive) disease [3]. Research has since demonstrated that axonal and neuronal loss actually begins at the earliest stages of the disease process, resulting in cognitive impairment and other early disability [4–8]. In addition, it is possible for patients with a more progressive clinical phenotype to have evidence of ongoing inflammatory activity either through clinical relapses or new MRI lesions. These distinctions are important on the clinical level as they influence treatment considerations, payer reimbursement, and eligibility for clinical trials. The classification scheme has therefore been recently revised to incorporate these principles [9**].
Classifications and diagnostic criteria
Relapsing-remitting multiple sclerosis
The vast majority of patients with multiple sclerosis initially follow a relapsing-remitting course, defined by acute exacerbations from which they typically completely or incompletely recover, with periods of relative clinical stability in between. An exacerbation, also referred to as a relapse or an attack, is defined by the International Panel on the Diagnosis of multiple sclerosis as ‘patient-reported symptoms or objectively observed signs typical of an acute inflammatory demyelinating event in the CNS, current or historical, with duration of at least 24 h, in the absence of fever or infection’ [10]. Diagnostic criteria have changed over time, based on the evolution of research and incorporation of technology to aid the diagnostic process, such as studies illustrating the use of MRI to improve diagnostic sensitivity without compromising specificity [11–14]. Application of the most current criteria, commonly referred to as ‘McDonald 2010’, does appear to result in earlier multiple sclerosis diagnosis compared with previous criteria [15].
Diagnostic criteria are based on a patient’s clinical presentation with typical symptoms and signs related to demyelinating lesions, usually accompanied by imaging that is consistent with multiple sclerosis, disseminated in both space and time. Common presenting syndromes include optic neuritis, sensory and/or motor manifestations of myelitis, and brainstem symptoms such as internuclear ophthalmoplegia. Presenting symptoms, the diagnostic process, and differential diagnosis are discussed in the following sections. The caveat to the application of the McDonald diagnostic criteria is that there must be ‘no better explanation’, meaning that the patient’s symptoms, signs, and imaging should not be diagnosed as multiple sclerosis if they are more consistent with an alternative disease process.
Dissemination in space (DIS) refers to the requirement that lesions affect at least two areas of the CNS typically affected by multiple sclerosis. This can be demonstrated clinically, such as in a patient with a prior history of optic neuritis who now presents with a brainstem syndrome. In this case, DIS is satisfied if there is objective clinical evidence of these two separate lesions or if there is objective clinical evidence of one lesion with a reasonable historical account of the other. However, often a patient will present after only a single event, termed a ‘clinically isolated syndrome’ (CIS). In this case, DIS may be satisfied if the clinician detects evidence for another separate lesion on neurological examination but may also be satisfied with clinical evidence for only one lesion by incorporating the patient’s MRI data. MRI criteria for DIS require the presence of at least one T2 lesion in at least two of the four areas of the CNS typically affected by multiple sclerosis: periventricular, juxtacortical, infratentorial, and spinal cord (Table 1). If the patient has a brainstem or spinal cord syndrome, the symptomatic lesion has presumably already been ‘counted’ and therefore does not count toward application of the MRI criteria for DIS.

Dissemination in time (DIT) refers to the requirement that CNS lesions have developed over time, to reduce the misdiagnosis of monophasic illness as multiple sclerosis. DIT can easily be satisfied clinically in a patient with two clinical attacks, again with objective clinical evidence for both attacks or for one with a reasonable historical account of the other. However, DIT can also be satisfied with a single clinical episode by the application of MRI criteria. On the patient’s initial MRI, DIT can be satisfied by demonstration of the presence of both gadolinium-enhancing and nonenhancing lesions on the same scan, as this illustrates that the lesions presumably developed at different points in time. Of note, the enhancing lesion may not be the symptomatic lesion, which has already been ‘counted’. In addition, DIT may be satisfied by the development of any new T2 and/or gadolinium-enhancing lesion with reference to the baseline scan, regardless of the time interval between them. MRI DIT criteria are shown in Table 2.

It should be noted that cerebrospinal fluid (CSF) analysis is not a requirement for the diagnosis of relapsing-remitting multiple sclerosis (RRMS) under McDonald 2010. However, it remains an important part of the evaluation for patients in whom the diagnosis is not entirely clear, either to provide support, paraclinical evidence for multiple sclerosis, or investigate other potential explanations for the patient’s presentation. Additional laboratory or other testing should be directed by the patient’s clinical presentation and whether there remains suspicion for a disease process other than multiple sclerosis. This is further discussed in the section on differential diagnosis.
Clinically isolated syndrome
The category of CIS was added to the new classification scheme, although the term has been in use for many years both in research and clinical practice. CIS represents a patient’s initial presentation with clinical symptoms typical for a demyelinating event. A patient is classified as having CIS when there is clinical evidence of a single exacerbation and the MRI does not fully meet RRMS criteria. From a practical standpoint, there is little distinction in the approach to a patient classified as having CIS compared with RRMS, as multiple studies have now demonstrated that patients with a typical CIS, especially those with brain lesions consistent with multiple sclerosis on MRI, have a high likelihood of going on to meet RRMS criteria in the future [16,17,18*] and early treatment is effective at preventing additional relapses [19–24]. The presence of oligoclonal bands seems to be important prognostically [18*,25], and several recent studies have suggested other potential CSF biomarkers as predictors of conversion from CIS to RRMS [26–29]. In addition, an inverse correlation has been noted between vitamin D level at the time of CIS and the likelihood of going on to meet RRMS criteria [30], but whether this is a marker for some other factor or this may be overcome with vitamin D supplementation is not yet known. Ocular coherence tomography (OCT) may also prove to be helpful as a predictor [31]. As additional research relating to predictive factors is completed, it can be incorporated into techniques such as machine-based learning that employs computerized classification algorithms to estimate future exacerbation risk [32]. This technique could potentially help stratify risk in CIS patients to aid the decision process regarding the timing of initiation of disease-modifying therapy.
Radiologically isolated syndrome
As MRI has become increasingly widespread for headache, trauma, and other conditions, abnormalities suggestive of multiple sclerosis have been noted in patients who have not previously experienced clinical symptoms of the disease. The term ‘radiologically isolated syndrome’ (RIS) was coined in 2009 [33] and has now been added to the revised multiple sclerosis classification scheme. The current formal diagnostic criteria for RIS are based on the initial 2009 publication, shown in Supplementary Table 1, http://links.lww.com/CONR/A31. They require that lesions are ovoid and well circumscribed, not consistent with a vascular pattern, and meet three out of four Barkhof criteria [34]: one gadolinium-enhancing lesion or at least nine total T2 lesions, one juxtacortical lesion, one infratentorial lesion, and three periventricular lesions. The findings must be incidental, meaning there must be no history of neurological symptoms suggestive of a demyelinating event and the lesions must not account for functional impairment. The lesions must not be better explained by a substance or toxic exposure or another disease process with a specific exclusion for those with extensive white matter disease not involving the corpus callosum. The criteria are likely to be updated in the near future to incorporate McDonald 2010 DIS principles.
In a recent study with an average of 4.4 years of follow-up, 34% of patients developed a first clinical event consistent with multiple sclerosis, although this does not represent the natural history of this classification as the group included patients treated with disease-modifying therapy [35*]. Younger age, male sex, and the presence of spinal cord lesions were noted to have predictive value. Owing to the lack of availability of evidence, currently there exists considerable variability in management, but many clinicians consider the presence of spinal cord lesions, whether the MRI is changing or lesions enhance with gadolinium to indicate ongoing disease activity, and/or the presence of oligoclonal bands in the CSF in the decision regarding whether to initiate disease-modifying therapy for multiple sclerosis in these patients.
Primary-progressive multiple sclerosis
The primary-progressive multiple sclerosis (PPMS) classification describes patients with progressive decline in neurological function from the time of disease onset. Patients most often present clinically with a progressive myelopathy although they may also present with a progressive cerebellar syndrome or other progressive symptoms as described further. McDonald 2010 criteria require at least 1 year of clinical disease progression as well as at least two of the following: evidence for DIS in the brain (at least one T2 lesion that is periventricular, juxtacortical, or infratentorial), evidence for DIS in the spinal cord (at least two T2 lesions in the cord), or positive CSF (isoelectric focusing of oligoclonal bands and/or elevated immunoglobulin G index). As in RRMS, symptomatic lesions are excluded from the MRI DIS lesion count. These are illustrated in Table 3 and the full McDonald 2010 criteria for both RRMS and PPMS are in Table 4.

Secondary-progressive multiple sclerosis
Secondary-progressive multiple sclerosis (SPMS), defined by gradual progression after an initial relapsing course, occurs in up to 40% of patients by 20 years after the initial event [36]. It is typically characterized by a gradual decline in neurologic functioning, often predominantly involving areas of the CNS previously involved during the relapsing course. The point of transition to SPMS can be difficult to define and is often recognized only in retrospect, at times years after subtle hints of progression first appear [37]. Research regarding potential imaging and laboratory biomarkers that distinguish SPMS from RRMS, better characterize the transition from RRMS to SPMS and even potentially predict the transition from RRMS to SPMS, is underway although each suggested biomarker currently requires further validation prior to clinical use [38–43].
Descriptive modifiers of multiple sclerosis phenotypes
Activity
The MS Phenotype Group recommends yearly assessment of clinical and brain MRI activity in patients with relapsing multiple sclerosis. Clinical activity is defined by relapses and brain MRI activity by the presence of gadolinium-enhancing lesions and/or new or unequivocally enlarging T2 lesions. Because spinal cord MRI activity correlates well with brain MRI activity [44], routine spinal cord MRI surveillance in the absence of clinical findings is not required for activity assessment. Regarding activity assessment in progressive patients, the Group recommends yearly clinical assessment, although they did not reach a consensus regarding imaging.
The modifier ‘active’ or ‘not active’ can be applied to each patient for the specified time interval of assessment. This allows for elimination of the previous classification of progressive-relapsing multiple sclerosis, which had described patients with progression from onset who also had evidence of inflammatory activity. Such patients can now be classified as PPMS active compared with those with a purely progressive course classified as PPMS not active. Patients who have not had a recent activity assessment can be classified with the modifier ‘activity not assessed’.
Progression
A diagnosis of progressive multiple sclerosis does not guarantee that the patient will continue to demonstrate ongoing decline. Some patients progress rapidly, some at a slow and steady rate, whereas others seem to reach a plateau [45]. This currently has implications for clinical trial enrolment, as clinical trials in progressive disease require documentation of recent progression for inclusion. In addition, it will hopefully have implications in the future regarding initiation of disease-modifying therapy for progressive disease and ongoing assessment of its effectiveness. The MS Phenotype Group therefore recommended a modifier regarding the current status of progression in patients with progressive disease, adding the term ‘progressing’ or ‘not progressing’ to modify the clinical phenotype. The lack of validated biomarkers for progression necessitate that this yearly assessment is purely clinical, based on patient-reported history and objective findings on clinical examination.
The application of the revised clinical classifications with modifiers is illustrated in Supplementary Figs. 1 and 2, http://links.lww.com/CO NR/A31.
SYMPTOMS AND SIGNS SUGGESTIVE OF DEMYELINATING DISEASE AND THEIR DIFFERENTIAL DIAGNOSIS
As described previously, the process of diagnosing multiple sclerosis generally begins with a patient who presents with the acute (relapsing) or insidious (progressive) onset of neurological symptoms. Prior to considering the application of multiple sclerosis diagnostic criteria, the clinician must determine whether the clinical history and examination, imaging, and other available data are consistent with demyelination related to multiple sclerosis. It should be noted that multiple sclerosis is not ‘a diagnosis of exclusion’ and therefore its diagnosis does not require an exhaustive search to exclude all other potential causes for the clinical presentation. Rather, the diagnosis is based on a constellation of findings that are typical for the disease, with tailored additional diagnostic workup required as an absolute only when atypical features are present. Typical presenting features, as well as those that should raise suspicion for an alternate disease process, termed ‘red flags’ are reviewed here. Examples are provided in the text and a more detailed list of clinical and imaging red flags developed by the Task Force on Differential Diagnosis in multiple sclerosis are presented in Supplementary Tables 2 and 3, http://links.lww.com/CONR/A31.
Clinical Features
Spinal cord syndrome
The most common clinical presentation of multiple sclerosis is symptomatology associated with acute onset of a partial transverse myelitis, typically sensory symptoms consistent with involvement of the dorsolateral cord [46*]. Depending on the extent of the lesion, symptoms may be unilateral or bilateral, at or below the level of the lesion, which in multiple sclerosis most commonly occurs in the cervical cord. The motor system as well as bladder and bowel function may be impaired. Acute complete transverse myelitis with resulting paraplegia is rare in multiple sclerosis and should prompt consideration of other disorders such as neuromyelitis optica spectrum disorder (NMOSD). Acute myelitis due to multiple sclerosis typically evolves over the course of days and begins to spontaneously recover over the course of a few weeks. Brain MRI is quite helpful as the majority of patients with a brain MRI suggestive of multiple sclerosis accompanying a partial acute transverse myelitis will go on to meet multiple sclerosis diagnostic criteria in the near future [47].
A more insidious onset should prompt consideration of PPMS, which in approximately 80% of cases presents as a progressive myelopathy [48]. In PPMS, motor symptoms such as weakness, spasticity, and difficulty with gait tend to predominate over sensory symptoms. Depending on the remainder of the clinical picture, consideration may also be given to compressive disease, toxic-metabolic causes such as B12 or copper deficiency, infection such as human T-cell lymphotropic virus, malignancy, or underlying genetic condition such as hereditary spastic paraparesis [46*,49*].
Other than the recommended brain and spinal cord MRI (discussed in further sections), the nature and extent of the diagnostic workup of a spinal cord syndrome should be driven by the clinical presentation. For example, a patient with a history of gastric bypass with insidious symptom onset will certainly require evaluation for vitamin deficiencies whereas a patient with an acute partial transverse myelitis that spontaneously improves as well as brain MRI suggestive of multiple sclerosis may need no further workup at all. Lumbar puncture is recommended in cases of progressive myelopathy in which multiple sclerosis is suspected given the special role CSF plays in the diagnostic criteria for PPMS.
A suggested algorithm for consideration of a spinal cord syndrome related to possible underlying multiple sclerosis is presented in Supplementary Fig. 3, http://links.lww.com/CONR/A31. Other reported potential causes of transverse myelitis are outlined in Supplementary Table 4, http://links.lww.com/CONR/A31, and of spastic paraparesis in Supplementary Table 5, http://links.lww.com/CONR/A31.
Optic neuritis
The differential diagnosis for suspected optic neuritis is quite broad and outlined in Supplementary Table 6, http://links.lww.com/CONR/A31; however, there are particular features that suggest multiple sclerosis-related optic neuritis as the cause as well as others that suggest alternative processes (Supplementary Table 7, http://links.lww.com/CONR/ A31) [50*]. Optic neuritis due to underlying multiple sclerosis typically presents with acute, unilateral, painful decrease in visual acuity that peaks within a few days and begins to recover within a few weeks [51]. A hyperacute presentation should raise suspicion for a vascular process, whereas a more insidious presentation should raise suspicion for an infiltrative disorder such as neurosarcoidosis, toxicmetabolic process such as B12 deficiency, or paraneoplastic syndrome although PPMS may rarely present with gradually worsening vision due to progressive optic neuropathy [48]. Simultaneous bilaterality is possible but uncommon and should raise suspicion for processes such as NMOSD, neurosarcoidosis, or Leber’s hereditary optic neuropathy (LHON), especially in the setting of a positive family history. In multiple sclerosis-related optic neuritis, pain with eye movements is typically present and mild to moderate in nature [51]. Painless visual loss should cue consideration of a vascular cause, especially in older patients, or LHON, whereas severe pain is more common in NMOSD. Phosphenes and scintillations may be present [50*].
Examination typically reveals impairments in acuity, low contrast vision, and color discrimination as well as an afferent pupillary defect [52]. Central scotoma is common and a variety of visual field defects are possible. Funduscopic examination is often normal but optic disc swelling may be seen [53]. Poor recovery, even without steroids, is uncommon in multiple sclerosis [52] and more suggestive of LHON or NMOSD [54]. Neuroophthalmology input, especially in cases in which red flags are present, is often helpful.
Brainstem or cerebellar syndrome
The most common brainstem presentation of multiple sclerosis is diplopia due to internuclear ophthalmoplegia, which may be bilateral, although diplopia may also result from a sixth nerve palsy [55]. Facial weakness or loss of sensation may accompany eye movement abnormalities or occur in isolation. Vertigo may occur due to a lesion anywhere along the vestibular pathways and ataxia may result from a cerebellar lesion [55]. Isolated trigeminal neuralgia as the sole presenting symptom of multiple sclerosis is uncommon. Third nerve palsy or complete ophthalmoplegia are more suggestive of other causes. Persistent hiccups, nausea, or vomiting are suggestive of area postrema lesion due to NMOSD. In relapsing multiple sclerosis as with other syndromes, the onset of brainstem or cerebellar symptoms is over hours to days; hyperacute onset is suggestive of a vascular cause especially if the symptoms are consistent with involvement of a clear vascular territory.
Approximately 15% of patients with PPMS will present with a progressive cerebellar or brainstem syndrome, characterized most prominently by gradually worsening ataxia [48]. They may also have progressive worsening of dysarthria, dysphagia, and, note, diplopia. MRI findings and the presence or absence of red flags will guide the differential diagnosis, which includes toxic/metabolic disturbances, malignancy, infiltrative processes, and others.
An algorithm for the evaluation of a patient presenting with an isolated brainstem syndrome is presented in Supplementary Fig. 4, http://links.lww.com/CONR/A31.
Cognitive impairment
Cognitive impairment is common in all multiple sclerosis phenotypes and begins early in the disease, although it is typically much more prominent in progressive than relapsing multiple sclerosis. Cognitive complaints often accompany other symptoms of multiple sclerosis and may help solidify a diagnosis; however, given their nonspecific nature and lack of association to a particular acute CNS lesion (at least with current standard imaging technique), cognitive symptoms alone without focal neurological symptoms are not usually helpful discriminators in the diagnostic process for RRMS.
PPMS may present with cognitive dysfunction without clear simultaneous focal neurological symptoms and signs, as recently illustrated in a small cohort of Italian patients [56*]. Several of these patients had atrophy and initially only nonspecific changes on MRI. They were only subsequently diagnosed with multiple sclerosis after additional testing such as advanced imaging with double inversion recovery revealing cortical lesions, or lumbar puncture was consistent with multiple sclerosis.
Other clinical presentations
There are several other less common clinical syndromes that may be consistent with a first presentation of multiple sclerosis. Cerebral hemisphere lesions, particularly large tumefactive brain lesions, can present as a hemispheric syndrome with symptoms that include aphasia, encephalopathy, and manifestations of increased intracranial pressure, in addition to motor and sensory symptoms. Paroxysmal symptoms are transient, recurrent, stereotyped symptoms such as vibrating or shock-like sensation with neck flexion (Lhermitte phenomenon), tonic spasms, trigeminal neuralgia, or paroxysmal dysarthria. Of note, for paroxysmal symptoms to be applied toward diagnostic criteria, they must be recurrent over at least 24 h. Other less common symptoms include seizures and symptoms related to disorders of thermoregulation or sleep [57] but these are rarely the sole presenting symptom of the disease.
Imaging features
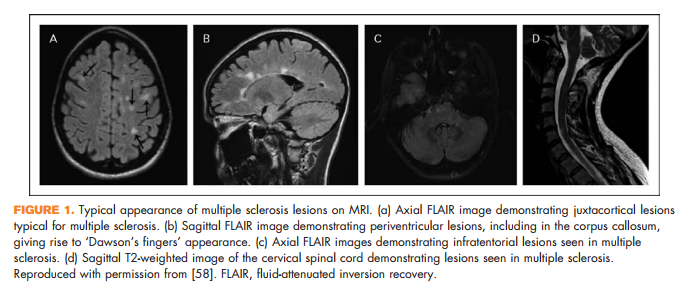
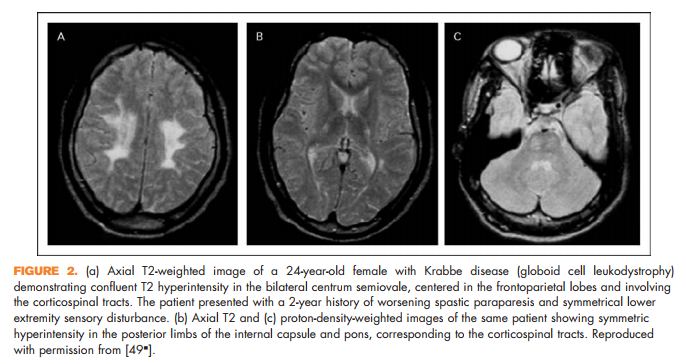
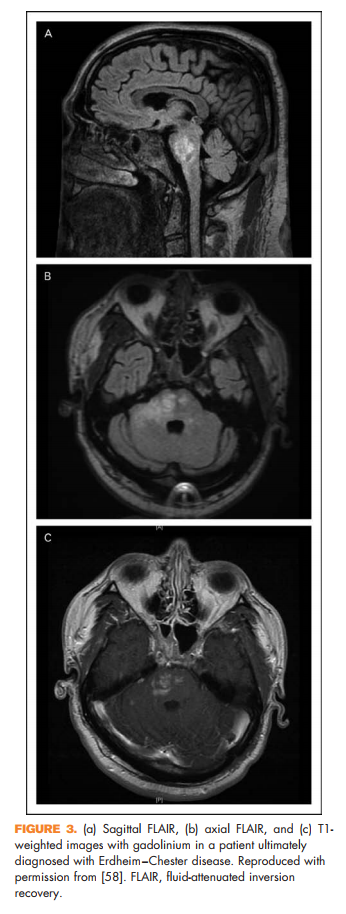
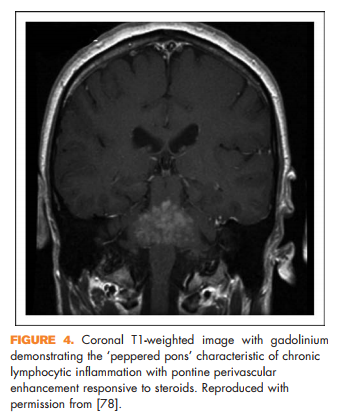
Brain lesions related to multiple sclerosis are typically ovoid, well circumscribed, oriented perpendicularly to the ventricles, and occur in characteristic locations: periventricular, juxtacortical, and infrantentorial (Fig. 1a–c) [58]. Spinal cord lesions are also well circumscribed, relatively small (typically two or less vertebral segments in length), occupy less than 50% of the cross-sectional cord area, and often involve the dorsolateral cord [59] (Fig. 1d). Lesions at least three vertebral segments in length are suggestive of NMOSD. Diffuse abnormalities in either the brain or spinal cord, especially in the setting of progressive symptom onset, should raise suspicion for a toxic/metabolic or genetic disorder (Fig. 2). New lesions typically enhance with gadolinium for approximately 6 weeks; enhancement that persists beyond 3 months should prompt consideration of other diagnoses such as sarcoidosis, histiocytosis (Fig. 3a–c), chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (Fig. 4), malignancy, or others relevant to the clinical context. Brain lesion load is on average, less in PPMS than RRMS, although atrophy accumulates more quickly in PPMS than RRMS. Supplementary Table 3, http://links.lww.com/CONR/ A31, outlines various imaging red flags and their differential diagnosis.
Differentiating multiple sclerosis from neuromyelitis optica spectrum disorder
One of the most common questions raised by referring physicians to multiple sclerosis specialists is whether a patient has multiple sclerosis or the much less common but potentially more severe inflammatory CNS disease NMOSD. The combination of typical clinical features for NMOSD and testing for antiaquaporin 4 antibodies (neuromyelitis optica immunoglobulin G) will correctly differentiate NMOSD from multiple sclerosis in many cases. However, the existence of seronegative NMOSD and overlap of certain clinical and imaging features with multiple sclerosis may complicate this distinction. Supplementary Table 8, http://links.lww.com/ CONR/A31, suggests differentiating features of the two disorders and they are reviewed here.
The cardinal clinical presentations of NMOSD are optic neuritis, longitudinally extensive myelitis, and hiccups/nausea/vomiting. Hiccups/nausea/ vomiting is rare enough in multiple sclerosis and common enough in NMOSD for this result in a correct NMOSD diagnosis once the symptoms have been recognized to be neurologic in etiology. Myelitis and optic neuritis, however, may present more of a challenge. Unlike in multiple sclerosis, wherein spinal cord lesions are usually small and discrete, in NMOSD, they are classically longitudinally extensive (at least three vertebral segments) and this can help differentiate the two disorders. However, a study from the Mayo clinic group demonstrated the possibility for ‘short transverse myelitis’ (less than three vertebral segments) in NMOSD, at times incorrectly leading to a diagnosis of multiple sclerosis until patients later presented with a more typical longitudinally extensive lesion or other classic feature of NMOSD [60*]. Central location of the spinal cord lesion and presence of tonic spasms helped distinguish NMOSD from multiple sclerosis and other disorders in certain cases in this study. Optic neuritis in NMOSD is more likely to be bilateral, posterior, and extensive including involvement of the chiasm [61,62], and result in poor recovery as compared with optic neuritis related to multiple sclerosis [63,64]. OCT may also help in differentiating the two; for example, NMOSD typically results in more retinal nerve fiber and ganglion cell layer thinning than multiple sclerosis, whereas subclinical abnormalities are commonly seen in multiple sclerosis but rare in NMOSD [65*].
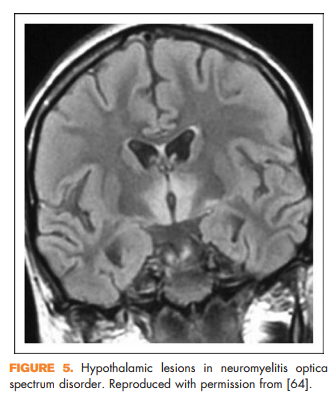
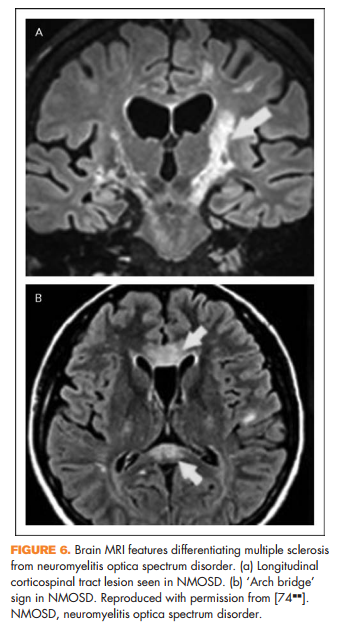
Other clinical features reported in NMOSD include focal brainstem syndromes, syndrome of inappropriate antidiuretic hormone secretion, narcolepsy, or other signs of hypothalamic involvement [66] (Fig. 5), and rarely myeloradiculitis [67] or myopathy with high creatine kinase [68,69], encephalopathy [70,71], or even hydrocephalus [72]. In addition, certain imaging features may help distinguish NMOSD from multiple sclerosis. It was previously thought that NMOSD did not result in abnormalities on brain MRI; however, it has subsequently been shown that patients with NMOSD can in fact have brain MRI abnormalities and some of these can resemble lesions seen in multiple sclerosis [73]. There are several features that may help distinguish them. Longitudinal corticospinal tract lesions (Fig. 6a), extensive hemispheric lesions, cervicomedullary junction lesions, bilateral symmetrical brainstem lesions, and periependymal lesions forming an ‘arch bridge’ (Fig. 6b) are more common in NMOSD whereas juxtacortical lesions, ovoid lesions perpendicular to the lateral ventricles (‘Dawson’s fingers’), and asymptomatic gadolinium-enhancing lesions seem to be more common in multiple sclerosis [74**].
Lumbar puncture may also be helpful. CSF in NMOSD often shows high white blood cell count, with neutrophils and eosinophils at times [75], compared with multiple sclerosis wherein more than 50 white blood cells are rare. Oligoclonal bands are highly associated with multiple sclerosis but are much less common in NMOSD [75,76]. Computeraided diagnosis employing methods for multimodal data fusion are also being explored and may prove to be powerful diagnostic tools [77].
CONCLUSION
As per 2013 revisions to multiple sclerosis phenotypic classifications, patients can be designated as having RRMS, CIS, RIS, PPMS, or SPMS. Modifiers regarding recent disease activity and progression have been added to further clarify current multiple sclerosis disease status.
The diagnostic criteria for RRMS and PPMS require presentation with a syndrome that is typical for demyelination, with demonstration of DIS and DIT. CIS requires a syndrome typical for demyelinating disease and RIS a typical MRI. When the clinical and imaging findings satisfy diagnostic requirements, the most crucial of which is that presenting features are consistent with a multiple sclerosis diagnosis, an exhaustive search regarding differential diagnosis is not necessary. Additional diagnostic testing should be tailored to the patient’s presentation, with particular attention to the presence of red flags that suggest a more appropriate alternate diagnosis.
Acknowledgements
None.
Financial support and sponsorship
The research of I.K.S. is supported by the National Multiple Sclerosis Society, the United States Department of Defense, and the Guthy Jackson Charitable Foundation.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
1. Belbasis L, Bellou V, Evangelou E, et al. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and metaanalyses. Lancet Neurol 2015; 14:263–273.
2. Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol 2014; 13:700–709.
3. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on clinical trials of new agents in multiple sclerosis. Neurology 1996; 46:907–911.
4. Kuceyeski AF, Vargas W, Dayan M, et al. Modeling the relationship among gray matter atrophy, abnormalities in connecting white matter, and cognitive performance in early multiple sclerosis. AJNR Am J Neuroradiol 2015; 36:702–709.
5. Rojas JI, Patrucco L, Miguez J, et al. Brain atrophy in radiologically isolated syndromes. J Neuroimaging 2015; 25:68–71.
6. Biberacher V, Boucard CC, Schmidt P, et al. Atrophy and structural variability of the upper cervical cord in early multiple sclerosis. Mult Scler 2014; doi: 1352458514546514. [Epub ahead of print]
7. Nygaard GO, Walhovd KB, Sowa P, et al. Cortical thickness and surface area relate to specific symptoms in early relapsing-remitting multiple sclerosis. Mult Scler 2015; 21:402–414.
8. Perez-Miralles F, Sastre-Garriga J, Tintore M, et al. Clinical impact of early brain atrophy in clinically isolated syndromes. Mult Scler 2013; 19:1878– 1886.
9. ** Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83:278–286. Recent revisions to multiple sclerosis phenotypic classifications by the MS Phenotype Group. This article outlines current classifications and recommends the use of modifiers regarding disease activity and progression.
10. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69:292–302.
11. Rovira A, Swanton J, Tintore M, et al. A single, early magnetic resonance imaging study in the diagnosis of multiple sclerosis. Arch Neurol 2009; 66:587–592.
12. Swanton JK, Rovira A, Tintore M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: A multicentre retrospective study. Lancet Neurol 2007; 6:677–686.
13. Montalban X, Tintore M, Swanton J, et al. MRI criteria for MS in patients with clinically isolated syndromes. Neurology 2010; 74:427–434.
14. Tur C, Tintore M, Rovira A, et al. Very early scans for demonstrating dissemination in time in multiple sclerosis. Mult Scler 2008; 14:631–635.
15. Brownlee WJ, Swanton JK, Altmann DR, et al. Earlier and more frequent diagnosis of multiple sclerosis using the McDonald criteria. J Neurol Neurosurg Psychiatry 2014; doi: jnnp-2014-308675. [Epub ahead of print]
16. Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology 2006; 67:968– 972.
17. O’Riordan JI, Thompson AJ, Kingsley DP, et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS. A 10-year follow-up. Brain 1998; 121 (Pt 3):495–503.
18. * Kuhle J, Disanto G, Dobson R, et al. Conversion from clinically isolated syndrome to multiple sclerosis: a large multicentre study. Mult Scler 2015; doi: 1352458514568827. [Epub ahead of print] A study of 1047 patients from 33 centers classified as CIS and followed for a median of 4.31 years, including CSF analysis, yielding significant findings regarding predictors of conversion to clinically definite RRMS.
19. Miller AE, Wolinsky JS, Kappos L, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol 2014; 13:977–986.
20. Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebocontrolled trial. Lancet 2009; 374:1503–1511.
21. Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet 2001; 357:1576–1582.
22. Filippi M, Rovaris M, Inglese M, et al. Interferon beta-1a for brain tissue loss in patients at presentation with syndromes suggestive of multiple sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet 2004; 364:1489– 1496.
23. Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet 2007; 370:389–397.
24. Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS study group. N Engl J Med 2000; 343:898–904.
25. Dobson R, Ramagopalan S, Davis A, Giovannoni G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: A meta-analysis of prevalence, prognosis and effect of latitude. J Neurol Neurosurg Psychiatry 2013; 84:909–914.
26. Rossi S, Motta C, Studer V, et al. Subclinical central inflammation is risk for RIS and CIS conversion to MS. Mult Scler 2015; doi: 1352458514564482. [Epub ahead of print]
27. Canto E, Tintore M, Villar L, et al. Validation of semaphorin 7A and ala-ss-hisdipeptidase as biomarkers associated with the conversion from clinically isolated syndrome to multiple sclerosis. J Neuroinflammation 2014; 11:181.
28. Villar LM, Espin˜o M, Costa-Frossard L, et al. High levels of cerebrospinal fluid free kappa chains predict conversion to multiple sclerosis. Clinica Chimica Acta 2012; 413:1813–1816.
29. Fialova´ L, Bartos A, Sˇ varcova´ J, et al. Serum and cerebrospinal fluid light neurofilaments and antibodies against them in clinically isolated syndrome and multiple sclerosis. J Neuroimmunol 2013; 262:113–120.
30. Martinelli V, Dalla Costa G, Colombo B, et al. Vitamin D levels and risk of multiple sclerosis in patients with clinically isolated syndromes. Mult Scler 2014; 20:147–155.
31. Perez-Rico C, Ayuso-Peralta L, Rubio-Perez L, et al. Evaluation of visual structural and functional factors that predict the development of multiple sclerosis in clinically isolated syndrome patients. Invest Ophthalmol Vis Sci 2014; 55:6127–6131.
32. Wottschel V, Alexander DC, Kwok PP, et al. Predicting outcome in clinically isolated syndrome using machine learning. Neuroimage Clin 2014; 7:281– 287.
33. Okuda DT, Mowry EM, Beheshtian A, et al. Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome. Neurology 2009; 72:800–805.
34. Barkhof F, Filippi M, Miller DH, et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997; 120 (Pt 11):2059–2069.
35. * Okuda DT, Siva A, Kantarci O, et al. Radiologically isolated syndrome: 5-year risk for an initial clinical event. PLoS One 2014; 9:e90509. Longitudinal follow-up of patients with RIS with risk factors for development of clinical symptoms and signs of multiple sclerosis.
36. Rovaris M, Confavreux C, Furlan R, et al. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol 2006; 5:343–354. 37. Sand IK, Krieger S, Farrell C, Miller AE. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler 2014; 20:1654–1657.
38. Pasquali L, Lucchesi C, Pecori C, et al. A clinical and laboratory study evaluating the profile of cytokine levels in relapsing remitting and secondary progressive multiple sclerosis. J Neuroimmunol 2015; 278:53–59.
39. Iwanowski P, Losy J. Immunological differences between classical phenothypes of multiple sclerosis. J Neurol Sci 2015; 349:10–14.
40. Dickens AM, Larkin JR, Griffin JL, et al. A type 2 biomarker separates relapsing-remitting from secondary progressive multiple sclerosis. Neurology 2014; 83:1492–1499.
41. Zastepa E, Fitz-Gerald L, Hallett M, et al. Naive CD4 T-cell activation identifies MS patients having rapid transition to progressive MS. Neurology 2014; 82:681–690.
42. Lavorgna L, Bonavita S, Ippolito D, et al. Clinical and magnetic resonance imaging predictors of disease progression in multiple sclerosis: a nine-year follow-up study. Mult Scler 2014; 20:220–226.
43. Paling D, Solanky BS, Riemer F, et al. Sodium accumulation is associated with disability and a progressive course in multiple sclerosis. Brain 2013; 136 (Pt 7):2305–2317.
44. Thorpe JW, Kidd D, Moseley IF, et al. Serial gadolinium-enhanced MRI of the brain and spinal cord in early relapsing-remitting multiple sclerosis. Neurology 1996; 46:373–378.
45. Pandey KS, Krieger SC, Farrell C. Clinical course in multiple sclerosis patients presenting with a history of progressive disease. Multiple Sclerosis Relat Dis 2014; 3:67–71.
46. & Cree BA. Acute inflammatory myelopathies. Handb Clin Neurol 2014; 122:613–667. Review of the differential diagnosis of acute inflammatory myelopathies.
47. Bourre B, Zephir H, Ongagna JC, et al. Long-term follow-up of acute partial transverse myelitis. Arch Neurol 2012; 69:357–362.
48. Miller DH, Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol 2007; 6:903–912.
49. & Weisfeld-Adams JD, Katz Sand IB, Honce JM, Lublin FD. Differential diagnosis of Mendelian and mitochondrial disorders in patients with suspected multiple sclerosis. Brain 2015; 138:517–539. Review of genetic conditions with features that may overlap with multiple sclerosis resulting in misdiagnosis.
50. & Petzold A, Wattjes MP, Costello F, et al. The investigation of acute optic neuritis: a review and proposed protocol. Nat Rev Neurol 2014; 10:447– 458. Review of the evaluation and differential diagnosis of potential acute optic neuritis.
51. The clinical profile of optic neuritis. experience of the optic neuritis treatment trial. optic neuritis study group. Arch Ophthalmol 1991; 109:1673– 1678.
52. Bermel RA, Balcer LJ. Optic neuritis and the evaluation of visual impairment in multiple sclerosis. Continuum (Minneap Minn) 2013; 19:1074–1086.
53. Hickman SJ, Dalton CM, Miller DH, Plant GT. Management of acute optic neuritis. Lancet 2002; 360:1953–1962.
54. Petzold A, Pittock S, Lennon V, et al. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry 2010; 81:109–111.
55. Miller DH, Chard DT, Ciccarelli O. Clinically isolated syndromes. Lancet Neurol 2012; 11:157–169.
56. & Calabrese M, Gajofatto A, Gobbin F, et al. Late-onset multiple sclerosis presenting with cognitive dysfunction and severe cortical/infratentorial atrophy. Mult Scler 2015; 21:580–589. Cognitive dysfunction as the initial presentation of multiple sclerosis.
57. Rae-Grant AD. Unusual symptoms and syndromes in multiple sclerosis. Continuum (Minneap Minn) 2013; 19:992–1006.
58. Katz Sand IB, Lublin FD. Diagnosis and differential diagnosis of multiple sclerosis. Continuum (Minneap Minn) 2013; 19:922–943.
59. Tartaglino LM, Friedman DP, Flanders AE, et al. Multiple sclerosis in the spinal cord: MR appearance and correlation with clinical parameters. Radiology 1995; 195:725–732.
60. & Flanagan EP, Weinshenker BG, Krecke KN, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015; 72:81–87. The occurrence of short-segment myelitis lesions in NMOSD.
61. Storoni M, Davagnanam I, Radon M, et al. Distinguishing optic neuritis in neuromyelitis optica spectrum disease from multiple sclerosis: a novel magnetic resonance imaging scoring system. J Neuroophthalmol 2013; 33:123– 127.
62. Khanna S, Sharma A, Huecker J, et al. Magnetic resonance imaging of optic neuritis in patients with neuromyelitis optica versus multiple sclerosis. J Neuroophthalmol 2012; 32:216–220.
63. Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012; 135 (Pt 6):1834– 1849. \
64. Fernandes DB, Ramos Rde I, Falcochio C, et al. Comparison of visual acuity and automated perimetry findings in patients with neuromyelitis optica or multiple sclerosis after single or multiple attacks of optic neuritis. J Neuroophthalmol 2012; 32:102–106.
65. & Bennett J, de Seze J, Lana-Peixoto M, et al. Neuromyelitis optica and multiple sclerosis: seeing differences through optical coherence tomography. Mult Scler 2015; doi: 1352458514567216. [Epub ahead of print] Differentiating multiple sclerosis from neuromyelitis optica through OCT and other neuroophthalmological techniques.
66. Viegas S, Weir A, Esiri M, et al. Symptomatic, radiological and pathological involvement of the hypothalamus in neuromyelitis optica. J Neurol Neurosurg Psychiatry 2009; 80:679–682.
67. Takai Y, Misu T, Nakashima I, et al. Two cases of lumbosacral myeloradiculitis with antiaquaporin-4 antibody. Neurology 2012; 79:1826– 1828.
68. Guo Y, Lennon VA, Popescu BF, et al. Autoimmune aquaporin-4 myopathy in neuromyelitis optica spectrum. JAMA Neurol 2014; 71: 1025–1029.
69. Suzuki N, Takahashi T, Aoki M, et al. Neuromyelitis optica preceded by hyperCKemia episode. Neurology 2010; 74:1543–1545.
70. Magana SM, Matiello M, Pittock SJ, et al. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 2009; 72:712–717.
71. Cheng C, Jiang Y, Chen X, et al. Clinical, radiographic characteristics and immunomodulating changes in neuromyelitis optica with extensive brain lesions. BMC Neurol 2013; 13:72.
72. Clardy SL, Lucchinetti CF, Krecke KN, et al. Hydrocephalus in neuromyelitis optica. Neurology 2014; 82:1841–1843.
73. Kim W, Kim SH, Huh SY, Kim HJ. Brain abnormalities in neuromyelitis optica spectrum disorder. Mult Scler Int 2012; 2012:735486.
74. && Huh SY, Min JH, Kim W, et al. The usefulness of brain MRI at onset in the differentiation of multiple sclerosis and seropositive neuromyelitis optica spectrum disorders. Mult Scler 2014; 20:695–704. Features of brain MRI that differentiate multiple sclerosis from NMOSD.
75. Jarius S, Paul F, Franciotta D, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 2011; 306:82–90.
76. Ebers GC, Paty DW. CSF electrophoresis in one thousand patients. Can J Neurol Sci 1980; 7:275–280.
77. Eshaghi A, Riyahi-Alam S, Saeedi R, et al. Classification algorithms with multimodal data fusion could accurately distinguish neuromyelitis optica from multiple sclerosis. Neuroimage Clin 2015; 7:306–314.
78. Pittock SJ, Debruyne J, Krecke KN, et al. Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain 2010; 133:2626–2634.
